发布日期:2018-10-18 16:24 来源:活性炭网 作者:活性炭网 浏览数:
内容概要 通常,活性碳是由木材,Ya和碳化和活化各种有机原料,如贝壳和煤炭生产。 碳化后通常在原料,氢,氧和碳在400〜700℃下加热,在惰性气体气氛下,除去挥发物的一部分(通
通常,活性碳是由木材,Ya和碳化和活化各种有机原料,如贝壳和煤炭生产。碳化后通常在原料,氢,氧和碳在400〜700℃下加热,在惰性气体气氛下,除去挥发物的一部分(通常是残留挥发物含量20-35%),由此制备适于活化的碳化物。激活是在碳化工序中产生,在高温下的碳化物600〜1000℃的水蒸汽的挥发性组分,二氧化碳气体与空气在碳化物或气化个碳原子的反应主要在10〜100埃的微孔结构,使内表面的侧面为1000m 2 / g以上。通过该方法,可以获得具有优异吸附性能的活性炭。活性炭的内部表面积,细孔分布,通过单独非常不同的材料类型的各种吸附性能不在设定保持时间所谓的热历史条件和炭化温度的加热速率,活化时间在每个活化温度,压缩气体的浓度等。为了在一个温度范围内均匀的进行这样的实验,装置能够反应的进行也是重要的。此外,特性的内表面积,例如在修改的日本药如活性炭(日本药典10)的第10列药用碳的性能,孔分布,亚甲基蓝吸附量,各种原料,因为它取决于等碘吸附从工业的观点来看,通过相同的设备和这些特性研究制造条件是非常有用的。
此外,在药用碳的日本药典10示出为细的活性炭,所述标准是非常严格和Hi戟和常规工业活性炭,上述唯一的相关仅吸附性能和各种特性的完全满意的标准我做不到。在这方面最重要的问题是4%或更小的药用碳的基准的点火残基,因此不符合标准无论多么优良吸附性。
目前,国内活性炭行业已成为严重的原料短缺。考虑到这些要点,在本研究中,我们选择未使用的原材料并考虑扩大材料选择范围。本研究的过程如框图所示。接下来,我们总结了获得的结果。
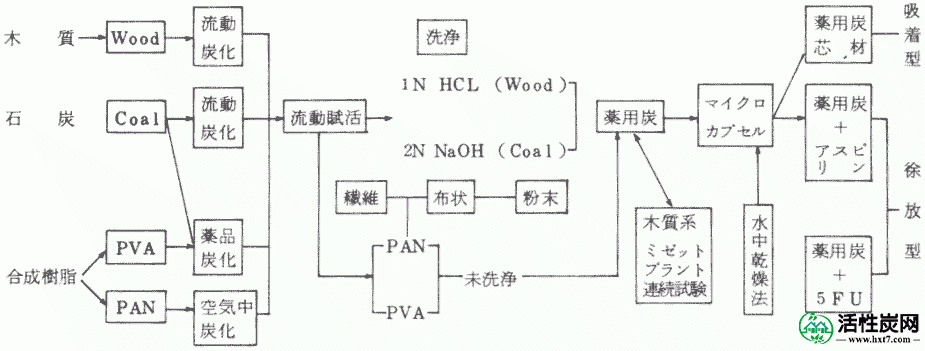
1。选择原料
选择煤,合成树脂和木质系统。选择的原因是煤可以稳定地供应大量储备。由于合成树脂具有很小的点火残基,有可能容纳的4%的参考或更少的点火残留物含量在药用碳的基础上,还木材是针对菲树木未使用的,木材也是唯一可以在自然界中再生的资源,并且推测它是一种有用的原料,具有低热残留。
2。碳化
B)木材和煤系统通过原型流化碳化炉碳化。该方法是一种资源节约型碳化方法,其可以仅通过样品中在200至600℃下产生的低分子量物质与用于流化气体的空气中的氧气之间的反应热来碳化。
B)在合成树脂体系中,我们发现了一种新的碳化方法,即当用浓硫酸处理聚乙烯醇时,可以高产率地制备碳化物约15倍的碳化物。而且,当用这种方法处理煤时,由于与流化碳化炉中制备的碳化物相比,强热残余物减少了约1/5至1/8,因此得到了适于生产药用炭的碳化物。还发现碳化物是在聚丙烯腈共聚物的空气中制备的。
3。激活
B)通过各种方法制备的所有碳化物通过间歇式流体活化装置活化。间歇式流体活化装置是透明的石英反应管,其允许直接观察高温流化状态,从而在均匀的温度下制备均匀的活化产物。因此,它是制造严格药用煤的合适方法。
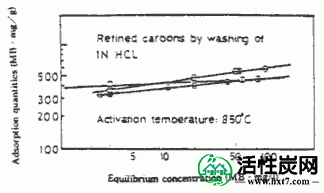
B)亚甲基蓝吸附由相应的碳化物的活化的性能,内表面区域具有市政版本产品或性能。该木基的活化是1.0N盐酸中,煤被发现能适应的标准的所有药用碳,并用Kaseisoda活化洗涤比由硫酸处理制备2.0N硬质合金更多。
C)的关系亚甲基蓝吸附量,并从碳化物的线性关系被激活的材料的内表面面积被建立,这取决于煤的煤的类型的直线的斜率也表明不同的线性一样。所有的木质系统都汇聚成一条直线。该结果表明,原料的选择比碳化和活化条件更重要。
D)来自聚乙烯醇和聚丙烯腈的共聚物的碳化物的活化产物不需要洗涤并且适合于药用煤的标准。此外,可以提及具有少量强热残余物的定性原料作为药用煤生产方法的基本条件之一。
4。适用于高级活性炭
通过水中干燥法的胶囊芯材料的吸附性能与壁乙基纤维素b)中的药用碳作为芯材的膜,它表示的亚甲基蓝吸附表面或内表面区域的约70%的吸附能力,而在水中的破损率经过分析,流出几乎没有得到药用碳的细粉末的,未来,有用的知识,包括使用这样的辅助吸附剂的用于人工肾脏作为包封胶囊。
B)胶囊吸附阿司匹林药用炭,更快的乙基纤维素浓度的在PH2的溶解试验的洗脱时间下〜12,如果PH增加阿司匹林变大洗脱水杨酸的。洗脱体积是服用未来时吸附在核材料的量的20-23%是这样的剂型的副作用阿司匹林,获得知识。
我的实验方法
1。原料
三种原料是木质,煤和合成树脂。木质材料使用9种菲律宾树和日本松树作为粉末活性炭的原料进行比较。与削片机中研磨树木,水含量在又105℃将粉碎的空气干燥器破碎机干燥至10%或更低,优选具有至2.0㎜筛分至样品的0.2的粒度。表I-1显示了这些工业分析值。
煤基接下来,北海道生产的煤化程度不同的太平洋煤,砂川三井煤炭(8日上煤),Horonai木炭的样品(TA在后面,SN,简称H)和印尼Baidouri炭(Bidoli)和下面干燥2.0㎜粒径(以下下文IB)的水分含量至约5%,在旋转干燥器。也有人在实验中使用太平洋炭置于四氯化碳和丙酮比重1.36的混合物后过筛,0.1〜1.0㎜其浮选煤(TAA的简称)。表I-2,3示出了工业分析值。
使用两种类型的合成树脂,聚乙烯醇和聚丙烯腈共聚物。即,使用日本合成化学工业有限公司制造,商品名Gosenoru(以下PVA的简称)(皂化(摩尔%度)或)的六个不同的样品的聚合度的聚乙烯醇。表I-4显示了每种PVA的工业分析值。聚丙烯腈共聚物(缩写为PANW)由Toho Beslon Corporation提供,但含量未知。该工业分析值如表I-5所示
| 样本 | 水分(%) | 灰(%) | 挥发物(%) | 固定碳(%) |
| Apitong | 9.71 | 0.98 | 83.71 | 5.64 |
| Mayapis | 12.48 | 0.03 | 80.01 | 7.34 |
| Tangile | 9.97 | 0.28 | 85.24 | 4.51 |
| Palosapis | 9.41 | 0.41 | 85.89 | 3.79 |
| Malabayabas | 11.16 | 0.41 | 77.72 | 10.71 |
| K. Bangkal | 11.68 | 0.76 | 77.18 | 10.37 |
| Ipil-ipil | 21.07 | 0.28 | 71.18 | 7.74 |
| Kakauate | 10.72 | 0.87 | 75.92 | 12.49 |
| 椰壳纤维,灰尘 | 15.93 | 6.44 | 53.97 | 23.66 |
| 松 | 12.00 | 0.40 | 37.00 | 50.57 |
| 样本 | 缩略语 | 水分(%) | 灰(%) | 挥发物(%) | 按钮编号 |
| TAIHEIYO | TA | 5.3 | 7.90 | 45.60 | - |
| Horonai | ^ h | 2.59 | 5.49 | 46.70 | - |
| 砂川 | SN | 3.10 | 6.70 | 42.20 | 5.6 |
| 样本 | 缩略语 | 水分(%) | 灰(%) | 挥发物(%) | 固定碳(%) |
| TAIHEIYO | TA | 5.5 | 10.72 | 46.13 | 37.65 |
| TAIHEIYO | TAA | 5.5 | 5.34 | 49.30 | 39.36 |
| Bidoli | IB | 11.7 | 3.31 | 41.95 | 43.04 |
| 样本 | 聚合度(摩尔%) | 分子量 | 灰分(wt%) |
| NH-18 | 99.7 | 1700 | 1.0 < |
| C-500 | 99.3 | 1720 | 1.0 < |
| KH-17 | 96.3 | 1680 | 1.0 < |
| NL-05 | 87.8 | 520 | 1.0 < |
| GH-17 | 80.0 | 1690 | 1.0 < |
| KL-05 | 80.0 | 490 | 1.0 < |
| 水分(%) | 灰(%) | 挥发物(%) | 固定碳(%) |
| 1.0 | - | 59.80 | 39.20 |
2。碳化
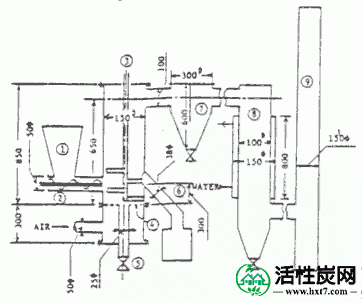
作为木质和煤的碳化装置,使用原型搅拌流化碳化炉。图I-1显示了轮廓。炉体
它由不锈钢制成,柱高和柱直径分别为650 mm和150 mm。在炉子的中心有一个桨式搅拌器,在流化床内以10rpm的转速搅拌。用螺旋进料器将样品连续送入炉中。根据炉温自动控制进料速率。在样品供应碗的正下方,放置一个埋头孔,其孔的直径为2mm,开口率为11.5%,通过该埋头孔将空气送入炉中。流化率基于样品的Umf(最小流化速度)预先设定。通过CA热电偶记录炉中6个位置的温度。流化床中心的热电偶连接到温度控制装置,以便调节螺旋送料器的旋转速度,使得该部分的温度恒定。电加热器缠绕在炉子的外表面上,并用于预热空气和样品。
在碳化方法中,将样品送入炉中,使得样品高度预先为柱的内径的1.5至2.0倍,吹入空气,并在使样品流动的同时用预热加热器加热样品。当流化床的内部温度超过200℃时,样品热分解并且温度开始升高;当温度超过400℃时,即使预热加热器停止,碳化也通过样品的氧化热进行。当系统达到这种状态时,将样品连续加入床中并碳化。从溢流管连续取出碳化物。根据经验,流化空气速度约为Umf的两倍,在该实验中,设定为6.0至9.0cm / sec。
由于通过上述碳化方法的低碳化产率,碳化方法不充分,PVA和PANW在硫酸或空气中碳化。
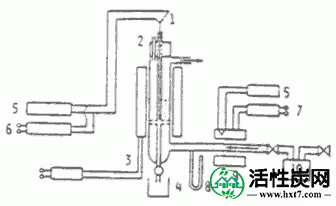
3。激活
活化装置是间歇式流化压块炉,是一种内径为40mm,高度为600mm的透明石英管,中心有石英分散板。分散板的孔径为1.0mm,开口率为10%。使用外部加热器进行温度控制。图I-2显示了它的华丽。活化温度为85.0℃,活化气体为过热蒸汽,每次活化100ml碳化物(粒径0.5-1.0mm)。因此,水蒸气也可用作碳化物的流化气体。蒸汽用计量泵将一定量的纯水送到加热器,蒸发并送入炉内。至于激活方法,首先从上部样品端口加入碳化物。随后,从设备的下部引入氮气,并调节其流速以使碳化物保持流动状态。接下来,通过温度控制器将碳化物在约30分钟内加热到预定的活化温度,此时,流化的气体立即转变成过热蒸汽。在反应完成后,通过再次切换到氮气并拉起从顶部插入的热电偶保护管,可以在下部样品接收器中获得活化材料。
激活如上所述的气体,有必要确定用于使用蒸汽的碳化物的流化气体的流速。在该实验中,碳化物体积为100ml时,每次,在该量发生吹气体,不能准确地从所述气体流率之间的流化层的压力损失之间的关系测定的碳化物的最小流化速度的U mf。因此,反应管以观察碳化物的流动状态直接透明石英管中,使用其中的为约1.5倍,该流化床高度的流速。如此确定流速为8.4㎝/秒。
4。清洁和
将活化剂研磨至-200目或更小,称重3g。然后,分别加入45ml的0.25,0.5,1.5和2.0盐酸,硝酸和硫酸,并将混合物在磁力搅拌器上在50℃下以70rpm搅拌10小时。通过布氏漏斗分离活性炭,并用40至50℃的纯净水洗涤直至滤液变为中性。将纯化的活性炭在110±3℃的干燥器中干燥5小时。
5。物性测试
(1)工业分析
根据日本工业标准JISM-8812的煤的试验方法测量原料和碳化物的灰分含量,挥发物含量,水分含量和固定碳含量。体积密度的测量根据JIS K-1474。
(2)亚甲蓝吸附量(MB)
的亚甲基蓝吸附量测定量为0.2g称重粉碎样品-300Mesh。置于锥形瓶中用旋塞,亚甲基蓝的标准溶液(MB300㎎/升)250毫升加入振荡30分钟(290转)后,在665nm下的亚甲基蓝浓度,用分光光度计的上清液,亚甲基蓝吸附量来测量相对于该样品1克以mg由计算确定的“/ G”表示。
(3)内表面积(S)
通过测定氮气在液氮温度下的吸附等温线,由BET方程确定内表面积。
(4)孔分布(10至100Å)
孔隙分数使用卡罗Erba的有限SORB制备MATIC 1800,表示从由克兰斯顿Inkly的方法10〜100埃的半径的孔分布从以ml / G”获得的氮的吸附等温线的孔体积。
(5)孔分布(75至75,000Å)
通过水银孔隙率测定法测定75至75,000的孔分布。
(6)碘吸附
称取0.2g活性炭,加入10ml 5%盐酸煮沸2分钟。冷却后,将碘试验溶液后(通过溶解K12.0g纯化水100毫升,随后升2加入12.7克”,纯净水和1L)中加入50mL,在滤液20毫升摇动5分钟,将淀粉后过滤它逐滴加入,并用硫代硫酸钠标准溶液的1/10ñ滴定,并计算的碘吸附为以mg / g表示。
(7)药用煤试验
根据日本药典10的药用煤试验方法进行通过酸洗纯化的活性炭的性能试验。
二木基原料
1。碳化物和碳化物的物理性质
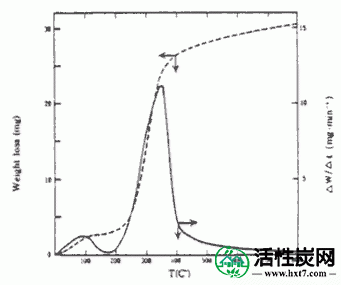
推断最佳碳化温度在碳化炉通过热天平,以获得所有样品的TG曲线。一个例子如图II-1所示。通常,首先,原料中的水分在100℃下作为峰解吸。然后,它与最大在约350℃,得到尖锐的曲线,该曲线的面积主要是相当于低分子量化合物的挥发物含量。甚至在煤类型中也获得这种TG曲线,并且挥发物含量被称为初级焦油。初级焦油与在该解吸温度附近氧气剧烈反应,氧的供给点火,并在充分的情况下燃烧。但不足以点燃减少供给量,由于通过Hatsujuku部分燃烧可以使用该热量来碳化,或空气的碳化温度供给量通过控制采样的供给量保持恒定你可以。当执行连续碳化基于此,碳化温度挥发性组分释放几乎去,即超过图略的峰值它是适当的。因此,流化碳化炉中的碳化温度从TG曲线设定为430℃。
在该实验中,进料空气流速保持恒定,控制样品进料速率并设定碳化温度。进给空气速度6〜9㎝/秒,将样品供给量在7〜8㎏“/小时调节至碳化温度炉温度设定,在±2变化范围保持 - 3℃。我成功了 表II-1显示了每种样品的碳化条件,碳化物产率,碳化时间和密度,表II-2显示了每种碳化物的分析值。
| Malabayabas | 430 | 8 | 10.8 | 1.50 | 20.9 | 26 | 0.12 |
| Kakauate | 430 | 8 | 12.3 | 1.48 | 25.0 | 18 | 0.14 |
| Bakauan | 430 | 8 | 10.8 | 1.50 | 14.0 | 26 | 0.12 |
| CAIR尘土 | 420 | 6 | 5.2 | 1.70 | 35.6 | 7 | 0.07 |
| Ipil-ipil | 430 | 7 | 11.3 | 1.60 | 34.0 | 22 | 0.18 |
| Mayapis | 430 | 6 | 10.5 | 1.46 | 23.3 | 19 | 0.16 |
| K. Bangkal | 430 | 8 | 10.8 | 1.50 | 14.0 | 24 | 0.11 |
| Apitong | 430 | 6 | 7.7 | 0.68 | 27.8 | 24 | 0.12 |
| Palosapis | 430 | 14 | 17.8 | 1.86 | 10.4 | 12 | 0.08 |
| Tangile | 430 | 五 | 4.36 | 0.62 | 34.2 | 16 | 0.07 |
| 松 | 430 | - | - | - | 27.0 | - | 0.12 |
| 样本 | 水分(%) | 灰(%) | 挥发物(%) | 固定碳(%) |
| Malabayabas | 5.00 | 2.40 | 10.20 | 82.40 |
| Kakauate | 3.30 | 3.80 | 14.40 | 78.50 |
| Bakauan | 2.20 | 5.10 | 20.30 | 72.40 |
| 椰壳纤维,灰尘 | 6.60 | 9.60 | 31.00 | 52.80 |
| Ipil-ipil | 2.20 | 4.70 | 24.12 | 68.98 |
| Mayapis | 1.18 | 1.56 | 16.01 | 81.25 |
| K. Bangkal | 1.07 | 6.60 | 19.18 | 73.15 |
| Apitong | 1.81 | 1.00 | 34.00 | 63.19 |
| Palosapis | 7.28 | 3.87 | 18.75 | 70.10 |
| Tangile | 9.42 | 1.46 | 24.90 | 64.22 |
| 松 | 2.41 | 1.80 | 47.81 | 47.97 |
2。活化粗产物的活化条件和性能
重量损失的表观速度是不理想的地区等也降低了产率,并且也是一个内部表面积增加而减小,但质量活化是在800℃或不能获得较少随着活化温度时,也900℃或更高它是活化温度估计,850℃〜900℃,因为在预实验中观察到的,我们为了获得最佳的温度进行研究,在850℃和900℃活化测试。结果示于表II-3。该表显示出在(MB)值的活化的产率(碳化物标准%)激活时间300㎎“/ G”,在活化温度(S)任何观察到的差异显著(MB)因为它不是在850℃活化。
| 样本 | 温度(°C) | 停留时间(分钟) | 收益率(%) |
| Apitong | 850 | 22.5 | 34.0 |
| 900 | 11.5 | 30.0 | |
| Mayapis | 850 | 36.5 | 48.0 |
| 900 | 18.5 | 49.0 | |
| Tangile | 850 | 20.0 | 24.0 |
| 900 | 16.0 | 18.0 | |
| Palosapis | 850 | 20.0 | 22.0 |
| 900 | 10.0 | 35.0 | |
| Malabayabas | 850 | 38.0 | 45.0 |
| 900 | 25.0 | 42.0 | |
| K. Bangkal | 8507 | 22.5 | 40.0 |
| 900 | 14.0 | 37.0 | |
| Bakauan | 850 | 35.0 | 39.0 |
| 900 | 24.0 | 41.5 | |
| Ipil-ipil | 850 | 45.0 | 41.5 |
| 900 | 25.0 | 44.0 | |
| Kakauate | 850 | 52.0 | 41.5 |
| 900 | 38.0 | 45.0 | |
| 椰壳纤维,灰尘 | 850 | 16.0 | 35.0 |
| 900 | -0 | - |
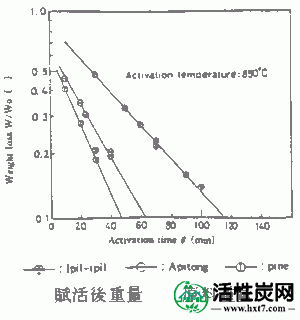
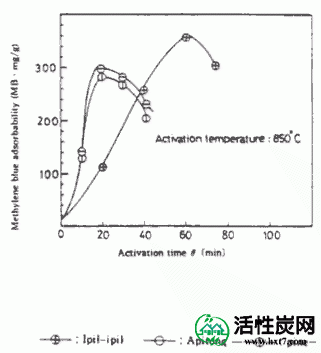
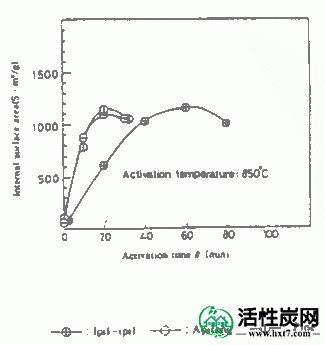
对于1pil-ipil如图典型实例II-2,3,4-(IP)Apitong(放C)碳化硅(AP)和派恩(PA),启动的时间和Mitsurukatsu产物产率(把A)率,显示了亚甲基蓝吸附(MB)和内表面面积(S)之间的关系。
3。清洁活化材料
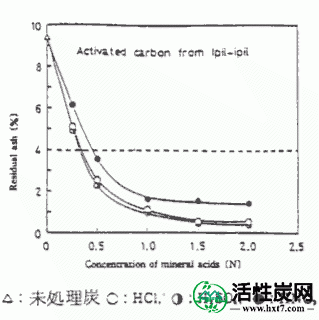
通过执行根据(4 I)的活化产物的酸洗涤纯化,任选地在寻求适用于4%或更低的洗涤条件点火残余物在日本药典10所示的药用碳的标准。图II-5显示了作为代表性实例的Ipil-ipil活化产物的酸浓度和纯化煤的热残余物残余物之间的关系。随着酸浓度增加,强热残余物被破坏,除了1.54%wt的硝酸处理高于1.0N,另一个是约0.5%wt。结果,洗涤酸浓度适当地为1.0N,并且使用廉价的盐酸更有效。Yaya的所有样品在上述条件下也符合日本局10的标准。
接着IPCA,粉碎APCA和后PACA活性炭用1.0N盐酸-200目纯化,在110±3℃下干燥3小时.. 0.02“称重和浓度300㎎”〜1.0克加入/ L”亚甲基蓝溶液200300500毫升后,和振动筛的旋转速度进行4小时,同时以290rpm振摇进行(垂直摇动)平衡吸附。用离心机分离活性炭后,测定上清液中残留的亚甲蓝浓度(mg / l)。图II-6显示了亚甲蓝的残留浓度(mg / l)与亚甲蓝吸附量(mg / l)之间的关系。结果发现,活性炭从低浓度区域到高浓度区域的吸附性优异。
4。纯化活性炭作为药用炭的评价
PAC代表性实例,APC Oyoo IPC活化温度850℃,以调整的激活在激活时间20,30,50分钟,并通过核酸浓度1.0N的纯化,是药用碳测试日台10 。因此纯化的煤相比,PH纯度试验是8.1至未处理的活性炭9.3被发现满足6.7到7.3,并使用参考任一酸。点火残基和加入未处理的活性炭物质在每个样品中尚未满足的标准,且足够满足PAC,APC和IPC 0.15〜0.57(百分比)减少到的程度的基准纯化。
据认为,南海材料未经处理的活性炭中酸性可溶物含量高的一个原因是钙含量高。氰化物,硫化物,硫酸盐,砷和重金属的含量均为未经处理的煤,符合标准。在吸附试验中,未处理的活性炭和纯化的活性炭之间没有观察到显着差异,并且发现满足所有标准。
III煤基材料
1。碳化物的制备和性质
通过(I)中所示的流化碳化装置以与木质体系相同的方式制备碳化物。除了褐色煤基,其他与粘合剂,Horonai木炭(H),砂川炭(SN)为原料的太平洋木炭(TA)碳化温度不能进入发送到设备用于在炉中的融合。因此,利用通过空气氧化降低粘性粘性的性质,采用以下方法。首先执行碳化物从室温至300℃,将其在460℃和430℃碳化在另一炉中冷却至室温,在室温下取出。根据该方法,不会发生由粘附引起的麻烦。表III-1显示了碳化物产率和工业分析值。根据该结果,产率在产率方面是有用的,而在木质系统中产率约为30%。由于煤而挥发木质原料是高约75%,由于产率是约45%,在碳化期间的脱挥发分挥发物含量为大概是因为木材是很多的。然而,对于药用煤标准而言重要的每个强热残余物都非常大,远远超过木材类型的1.0至4.7%的值。
| 样本 | 碳化温度 | 水分(%) | 灰(%) | 挥发物(%) | 固定碳(%) | 收益率(%) | 内表面积(m 2 / g) |
| TAIHEIYO | 600 | 2.25 | 16.15 | 17.27 | 64.38 | 47 | 51 |
| TAIHEIYO | 800 | 2.21 | 21.81 | 8.41 | 67.58 | 35 | 17 |
| Horonai | 460 | 4.19 | 6.70 | 28.75 | 60.36 | 63 | 23 |
| TAIHEIYO | 430 | 1.92 | 7.50 | 32.74 | 58.29 | 80 | 22 |
2。激活
活化装置和方法在(I)中所示的间歇式流动活化反应器中进行。通过直接观察透明石英管中碳化物的流动状态来测量活化炉中流化气体的流速,并将其作为当流化床的高度达到固定层高度的1.5倍时的流速。该值约为10至12厘米/秒。发现反应器中每种碳化物的流化气体的速度几乎相同,因此对于任何碳化物,活化时的蒸汽量几乎相同。
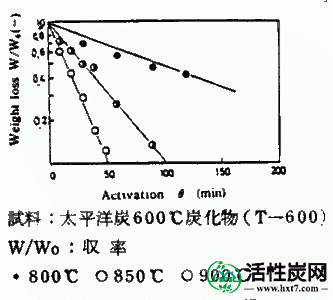
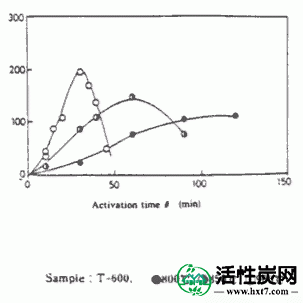
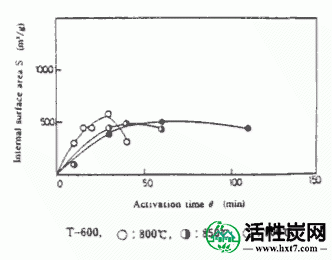
在活化温度800,850和900℃下的每种碳化物的活化时间(θ)与碳化物W / Wo( - )的重量损失率之间的关系的实例显示在图III-1中。在每个活化温度下活化时间(θ),亚甲蓝吸附量(MB)和内表面横向(S)之间的关系示于图III-2和III-3中。MB和S值都随着激活时间而增加并显示最大值,然后减小。活化剂的孔分布如图III-4所示。孔体积为0.8-2.0ml / g',是碳化物的1.5-3.0倍,超过商品0.38-0.6ml / g'。
3.评价为药用木炭
根据(I的4)进行每种活化物质的纯化,以研究符合药用煤标准的纯化条件。为了纯化,对活化材料中强热残余物的约20%或更少的活化时间获得的活性炭进行实验。用1.0-4.0N盐酸和苛性钠洗涤后,测定纯化活性炭的强热残余物。结果,两种活化剂都不符合日本药典10中所示的4%或更少的强热残余物。因此,从煤炭到药用炭的制造方法,需要设定活化条件以制备具有较少强烈的热残余和优异的吸附性能的活化物质,并进一步选择原料。
4。煤硫酸处理
因此,我们选择软灰含量较低的煤并尝试硫酸处理。第一(缩写为IB)印度尼西亚Bidoli煤作为原料如木炭太平(TAA)的重力分离进行实验。TAA示于表III-2,1B的碳化物,升温至170℃下用覆套式加热器置于浓硫酸样品的0.5先前粒径1.0㎜圆柱形可分离烧瓶巴尔具有1,000 ml的内部容积。在使用搅拌器以30rpm搅拌的同时缓慢加入样品。样品和1浓硫酸的重量比的量:1至1:7中进行的产物的产率几乎是恒定的1:实验在4进行。将生成的含有亚硫酸气体的气体用1.0N苛性钠水溶液处理。在一个实验中样品重量为100g'。反应温度为0.99℃时,在气体中不再产生时,观察到反应时间,TAA:5小时,IB:3.5小时(以下称产物TAAS,IBS略)的反应完成后,在容器中的产品冷却至室温,过滤,用纯化的温水洗涤直至溶液中和。
| 样本 | 缩略语 | 水分(%) | 灰(%) | 挥发物(%) | 固定碳(%) |
| TAIHEIYO | TA | 5.5 | 10.72 | 46.13 | 37.65 |
| TAIHEIYO | TAA | 5.4 | 5.34 | 49.30 | 39.36 |
| Biodoli | IB | 11.7 | 3.31 | 41.95 | 43.04 |
| 样本 | 缩略语 | 水分(%) | 灰(%) | 挥发物(%) | 固定碳(%) |
| TAIHEIYO | TAAS | 20.40 | 2.01 | 34.42 | 43.17 |
| Biodoli | IBS | 18.92 | 1.41 | 35.54 | 44.13 |
| Biodoli | IBC | 9.46 | 4.97 | 32.59 | 52.98 |
之后,将其在干燥器中在150℃下干燥12小时。此时TAAS和IBS的收率为97.8%,90.4%(wt)。表III-3显示了TAAS和IBS的工业分析值。这里要注意很重要的一点是,TAAS,下灼烧残渣含量在IBS 2.01,1.41%,这是已经获得了基于木材的碳化物优越药用碳性能灼烧残渣1.0 〜是一个值接近1.8%,因此在纸由煤得到的产物用浓硫酸猜到材料和适合于制备药用碳的堆肥反应。
TAAS,IBS和活化温度850℃的IBC,进行药用碳测试当天站10,用于获得在激活时间30,30,90min由2.0N盐酸或氢氧化钠溶液洗涤每个激活的对象的纯化的碳结果,PH为10至在每个原煤11,虽然碱度比木材稍高,每个纯化木炭6.4-6.8盐酸洗涤,通过与苛性钠溶液洗涤满足6.8-7.0,和两个标准我发现了。酸溶性级分是在每个原煤参考值大于3%,已经发现,以满足充分地或者与纯化洗涤的标准。对于未经处理的煤,强热残余物远高于标准的4%,但纯化的煤用盐酸洗涤,IBSA满足3.48%的标准。然而,TAASA和IBCA显示的值为4.54,9.62%,未达到标准。在用苛性钠溶液洗涤,TAASA和IBSA是TAASA与更多的氢氧化钠溶液2.0N洗涤,因为它是3.20,1.80%下IBCA4.74%,IBSA被发现满足标准。
酸处理的该方法中,在所得到的碳化物减少了对点火残余物至接近木材的值,和一个常规的碳化过程中热是不必要的,激活时间也被缩短,在经济上有用它是这样的方法。此外,活化剂的性能也很好,并且获得了从Bidoli煤炭太平洋煤等制备药用煤的前景。
IV合成高分子量原料
1。聚乙烯醇(PVA)的碳化
根据PVA的TG曲线,从约200℃发生急剧的重量减轻,并且在500至600℃下,其90%或更多通过热分解而挥发。曲线上600℃下的碳化物产率是4.3至9.0wt%的非常低的值。因此,发现通过简单地加热PVA,不可能以高产率获得作为活性炭原料的碳化物。因此,当添加各种化学品时从TG曲线获得的碳化产率示于表IV-1中。如上所述,化学添加的效果显着,特别是添加浓硫酸的效果显着,并且碳化产率是未添加的PVA的约15倍。
| 酸(5 wt%) | 碳化物产量(wt%) |
| 盐酸 | 19.02 |
| H 3 PO 4 | 27.30 |
| H 2 SO 4 | 63.10 |
| ZnCl 2 | 32.70 |
考虑到上述结果,使用用浓硫酸处理的相同设备使PVA和浓硫酸反应。首先,将浓硫酸置于内部容积为1,000ml的圆柱形可分离烧瓶中,并通过覆套式加热器加热至170℃。在使用搅拌器以30rpm搅拌的同时逐渐添加PVA,并且PVA与浓硫酸的重量比为1:1至1:7。将生成的含有亚硫酸气体的气体用1.0N苛性钠水溶液处理。尽管在反应时间内不再观察到上述气体的产生,但将该时间作为反应时间,因为所有样品约为5小时。反应完成后,将容器中的产物冷却至室温,过滤,并用温热的离子交换水洗涤直至滤液变为中性。之后,将其在干燥器中在150℃下干燥12小时。(此产品在下文中缩写为PVAS。)生物学上为黑色的PVAS,粒径为1.2-2.0mm,堆积密度为0.5-0.54g / ml。PVA的重量和浓硫酸1:1〜1:碳化物OsamuTsutomu 7是57〜62%(重量),其中,所述低浓硫酸为1:活性炭材料以1:1的重量比所得到的碳化物和。
| 样本 | 收益率(%) | 内表面积(m 2 / g) | 灰(%) | 挥发物(%) |
| 潘伟 | - | - | 0.007 | - |
| 潘阿 | 67.9 | 110 | 0.008 | 28.12 |
| 潘B | 64.3 | 130 | 0.009 | 19.00 |
2。聚丙烯腈(PAN)共聚物的热缩合
将纤维状层状聚丙烯腈(以下简称PANW)的共聚物切割成1.0~2.0mm后,通过以下方法进行缩合。即,插入件放置在金属丝网在(I)中所示的石英反应管间歇式流化床活化装置中,温度230℃的反应管,2小时和在空气中5小时的中心的样品10 G”并且假设空气量是自然通气(下文中产品缩写为PANA,PANB)。
PANA,B等的产率分析值示于表IV-2中。碳化物的产率接近于煤的产率,并且约为氯乙烯,聚碳酸酯树脂等的两倍。与PVA一样,该结果与煤等相比几乎没有强烈的热残余。它显示出比木质碳化物更低的值,并且获得用于制备药用炭的合适的碳化物。
3。激活和激活的特征
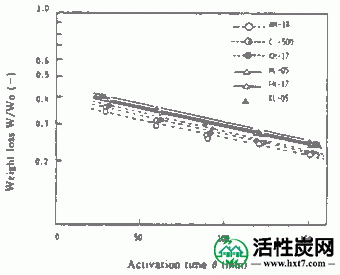
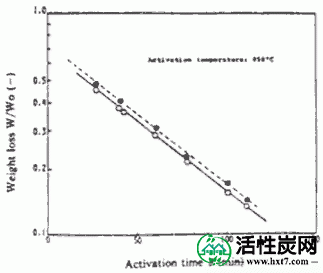
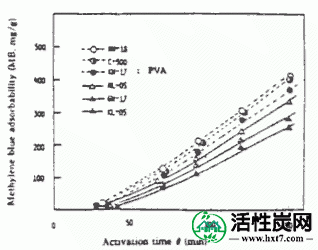
使用间歇式流化床活化装置中所示的PVA和碳化物PVAS和PANA从PANW和更早PANB,和水蒸汽以及制备煤木基并用850℃激活为活化气体。激活时间(THETA)和碳化物的重量损失,W /禾( - )。在图中所示的关系IV-1,IV-2。
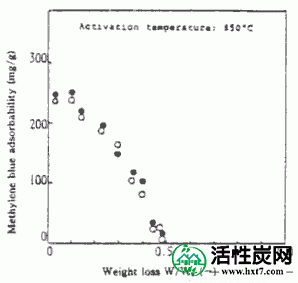
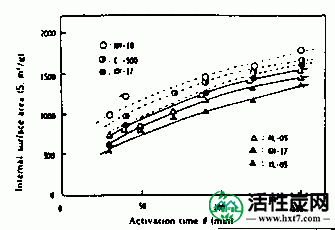
接着,在图IV-3 PANA,碳化物重量损失W /禾B在激活温床850℃ -表示亚甲基蓝吸附MB之间的关系()。随着W / Wo( - )下降,PANA和B都显示出增加MB的趋势。煤或木材的这种趋势是相同的。也显示激活时间(THETA)和Meterenburu吸附MB和在活化温度850℃对于每个PVAS图IV-4,IV-5的内表面面积S的关系。亚甲蓝吸附量MB和S均随着θ的增加而增加,在W / Wo( - )0.3下制备MB值为110至210mg / g'且S为1,000至1,500m 2 / g'的活化物质。是的。在该实验中,由NH-18制备的活化剂的性能非常好。
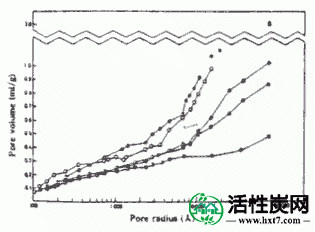
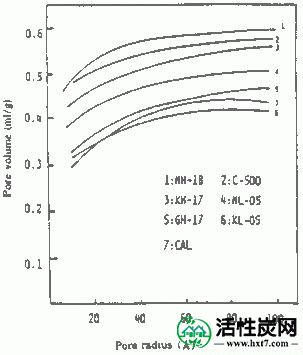
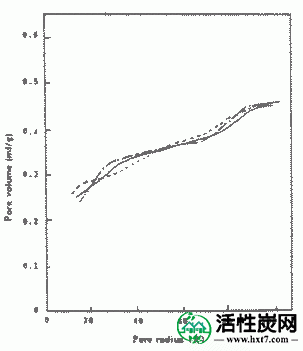
图IV-6和IV-7显示了从PVAS和PANW获得的活性炭的半径为10至100的孔体积的关系。从PVAS漂亮的孔隙体积活性炭是多方面的,10〜40A都相当大毛孔,PAN略有孔隙体积虽小,但在占有同样的方式毛孔多数在10〜40A。与用于比较的药用煤的结果相比,认识到它们与两者相当。
4.评价活性物质为药用炭
PANA,根据日本药典10 B中的药用碳测试和通过活化温度850℃下进行PVAS得到的活化产物的结果,在每个测试项目值满意的药用碳基准进行显示。由此,认为使用PANW和PVA作为原料的活性炭与木质和煤类型相当,并且可以制备药用炭而无需洗涤酸或碱。
| 样品\有机气体 | C 6 H 6 | C 4 H 9 SH | C 3 H 7 COOH | CH 3 NH 2 |
| PANAC | 38.63 | 260.58 | 35.48 | 37.35 |
| Y-20 | 52.55 | 62.19 | 28.16 | 51.34 |
| CAL | 50.28 | 46.83 | 24.38 | 44.36 |
5.特色用途
(1)PAN活性炭的有机气体的吸附
各种有机气体的吸附试验结果如表IV-3所示。实验中使用的PANAC在850℃下活化25分钟,S,640m 2 / g',M。B,65mg'/ g'。从烟煤活性炭椰子壳作为原料用于比较(Y-20),一直还描述活性炭(CAL),现在PANAC相对于吸附量丁基硫醇,可以看出,非常大。此外,PANAC是大量的科技酸,甲胺吸附的苯的吸附比市售活性炭小。顺便说一下,除了苯的50倍与吸附丁基梅尔卡丁烷(重量)样品,重复该操作五次过滤出从在室温下搅拌30分钟,2小时干燥苯在0.99℃后,再次测定丁基硫醇的吸附量时,吸附量为218.62%。这表明用丁基硫醇吸附的PANAC可以通过溶剂法再生。
PANAC吸附大量硫醇的原因被认为是PANAC中N - N氮的影响,因为PANP几乎没有吸附性能。
目前,由于作为难闻气味污染问题的硫醇没有特别优异的吸附剂,因此PANAC被认为是防止恶臭的有用材料。
(2)PAN纤维状活性炭
PANAC是纤维的PAN作为原料的形状,当然,可以由粉末形式,或颗粒有可能使模塑的纤维,毡,薄膜这样的。
目前,实际使用的清洁空气(特别是脱除硫醇的基本气味)和废水处理的目的良好的机械强度,能够制造大的毡制品的表面积。
类似于许多工业产品,PANAC的性能取决于其制造条件,但到目前为止获得的PANAC性能的一个例子列于表IV-4中。
(3)PVA活性炭的分子筛
平衡用中获得的产物的浓硫酸处理的饱和蒸气压的热处理在400〜1000℃的温度下在氮气流中,将得到的不同的目标分子吸附碳化物PVA大小这里从吸附量确定总孔体积,并与市售的分子筛power-Bon(商品名MSC5A,由Takeda Pharmaceutical Co.,Ltd。制造)比较。测试结果是表IV-5中的力。这表明PVAS在900至1000℃的加热温度下的孔结构类似于市售MSC的孔结构。根据上述结果,从硫酸处理过的聚乙烯醇炭中发现了分子碳生成的可能性。
| PANAC |
粒径 (mmΦ) |
体积密度 (g / cc) |
表面积 (m 2 / g) |
吸附量 | |||
|
碘 (mg / g) |
亚甲蓝 (ml / g) |
苯 (%) |
丁基硫醇 (%) |
||||
| 颗粒状A. | 0.330 | 580 | 960 | 70 | - | - | |
| 颗粒状B. | 0.270 | 850 | 1060 | 170 | - | - | |
| 颗粒状C. | 0.25到1.0 | 0.293 | 950 | 1110 | 189 | 34.6 | 340.0 |
| 纤维 | - | - | 1152 | 2850 | 258 | 43.6 | 1350.0 |
| 毡 | - | - | 1000 | 2600 | 258 | 49.8 | 4300.0 |
| 市售活性炭A. | 0.25至0.34 | 0.55 | 1100 | 1116 | 272 | 38.9 | 81.4 |
| 市售活性炭B. | 0.25到2.00 | 0.51 | 720 | 992 | 194 | 30.0 | 117.6 |
| 碳化温度(°C) | 苯 | 环己 | 四氯化碳 | 甲醇(ml / g) |
| 400 | 0.27 | 0.30 | 0.22 | 0.25 |
| 500 | 0.22 | 0.18 | 0.14 | 0.18 |
| 600 | 0.21 | 0.12 | 0.12 | 0.17 |
| 700 | 0.22 | 0.10 | 0.10 | 0.17 |
| 800 | 0.21 | 0.07 | 0.09 | 0.16 |
| 900 | 0.18 | 0.04 | 0.04 | 0.13 |
| 1000 | 0.19 | 0.04 | 0.02 | 0.13 |
| MSC | 0.16 | 0.04 | 0.07 | 0.15 |
V应用(1)药用木炭微胶囊
1。前言
药物检查微囊药用炭作为在医疗领域中这种使用的芯材。药用炭,活性炭布近年来污物去除异味卧床患者,活性碳纸,或住院治疗室空气净化,更寿命试验等使用椰壳活性炭作为辅助吸附剂人工肝。用涂有椰壳活性炭Koroshion膜吸附多个白蛋白微胶囊的那些,也已被释放,如球状活性炭用火棉胶膜碳石油涂敷沥青类原料跟着白蛋白处理胶囊剂,它们据说它大量进入体内并吸附并去除血液中的毒物。但是实施例中报道的详细的制备条件,并用粉末状的活性炭胶囊的性能是小的。作为芯材的药用碳这里,以溶解乙基纤维素(Etcell)的二氯甲烷绿泥石,则制备条件含薬滴加含水明胶溶液中,产生的胶囊的内表面面积的煤根据胶囊水干燥方法,亚甲基蓝吸附揭示考虑孔隙这样的厚度的分布和测量胶囊的性能。
2。实验
(1)实验方法和装置
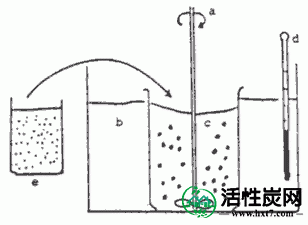
通过水包水干燥法进行包封。(II)在芯材中使用的药用碳,其中在良好PAAC(药用碳,其中松木为原料),将其粉碎至-300Mesh的特性的报道,其性能是亚甲基蓝吸附:240毫升/克,内表面积:800米2 / G”,孔体积:半径10〜100埃为0.34毫升/克。首先,药用碳20克“称重,以溶解乙基纤维素预先二氯甲烷250毫升。该溶液的Etcell浓度为0.4,0.9,11.2,1.8,3.6%。该%指示是(g / ml×100)。同样如下。然后20分钟并以杯的Nex塔拉剧烈搅拌下加入PAAC20g各浓度的乙基纤维素溶液。如图每乙基纤维素悬浮PAAC溶液中在40℃下置于恒温水浴中的容器滴加到4%的明胶水溶液千毫升。V-1。搅拌叶片逐渐蒸发在500〜800rpm下搅拌速度与叶片直径60㎜Fai四个叶片搅拌的同时,除去二氯甲烷。24小时后,取出胶囊,用40℃的纯化水用超声波洗涤器反复洗涤。胶囊用真空干燥机在27℃下干燥20小时后,进行在玻璃容器实验密封。
3。实验结果和讨论
(1)胶囊产量
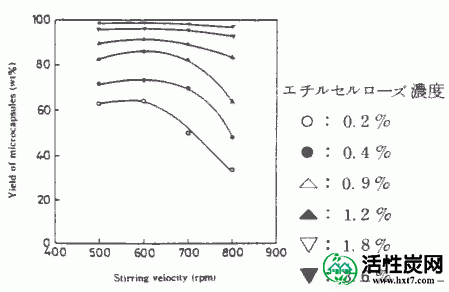
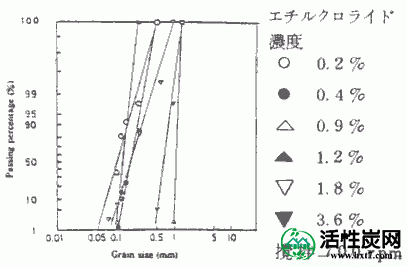
通过在上述方法中包封通过在Etcell浓度为0.2至3.6%的溶液中添加PAAC 17g'而制备的悬浮液。所得胶囊的收率与乙基纤维素浓度%的关系如图V-2所示。乙基纤维素浓度%越低,产率越低。当搅拌速度为600rpm或更高时,当乙基纤维素浓度低时,显示出快速降低的趋势。但是,在乙基纤维素浓度为1.2%或更高时,发现搅拌速度的影响小并且产率大致相同。接下来,胶囊的粒度分布曲线的实例显示在图V-3中。总体而言,均匀系数为1.1至1.5,并且在该制备条件下获得了良好的粒度分布。在这种情况下,胶囊的制备条件是搅拌速度为700rpm,乙基纤维素浓度为0.2-3.6%。
(2)胶囊性能
图V-4显示了当搅拌速度改变时乙基纤维素浓度与胶囊内表面积之间的关系。内表面积作为搅拌速度在每个乙基纤维素浓度的增加,合适的胶囊的制备条件被发现搅拌速度600〜800rpm的转速,是乙基纤维素浓度0.9%的逐渐增加。
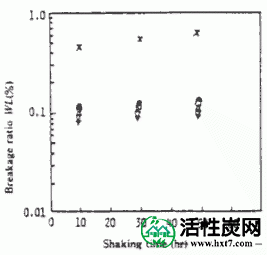
图V-5显示了在搅拌速度为700rpm的每种乙基纤维素浓度下获得的胶囊的失效率与摇动时间之间的关系。每个胶囊的失败率是非常小的,即,PAAC芯材几乎不流出,在比较中使用的商业的球状活性炭的故障率是在本研究中使用的胶囊的0.45至0.7%的失败率是非常发现它是一个很小的值。
通常,粉末和颗粒状活性炭具有严重的缺点,例如在使用过程中颗粒流出。在医疗领域中,例如,即使将其用作人造器官的辅助吸附剂,也存在消除该缺点的问题,并且认为封装是有用的手段之一。
使用药用炭作为芯材,研究了使用乙基纤维素作为壁膜的水中干燥法制备胶囊的条件和所得胶囊的性质。
结果,胶囊的最佳调节条件是壁膜的乙基纤维素浓度为0.9%,核心药用煤量为10至20g,搅拌速度为600至800rpm,胶囊产率为65至85wt%。产量下降得越快。内表面积和亚甲基蓝吸附从670胶囊700米的2 / G',194〜200毫克/ G“变得表明核心药用碳的性能的80%左右。半径为10至100的孔体积几乎与核心药用炭的孔体积相同。由于每个胶囊在水中进行摇动试验,认为几乎没有发现任何胶囊雾化的核心材料药用煤的流出,并且它被认为是粉末药用木炭造粒的有用手段。

VI应用(2)阿司匹林吸附煤微胶囊
1。前言
考虑到前一部分中获得的胶囊的调节条件,这里研究了从远古时代用作医疗护理的阿司匹林的包封。阿司匹林胶囊用乙基纤维素包裹在阿司匹林颗粒中。其目的是控制胃肠道的吸收率和预防消化组织的出血。此处被预吸附的阿司匹林药用碳,确定乙基纤维素和胶囊的调整状态的知识由于壁膜和在水中干燥法,经历使用胶囊阿司匹林的溶出试验中的缓释胶囊。
2。实验方法
(1)阿司匹林吸附药用炭的制备
阿司匹林粉末用作阿司匹林粉末。精确制备4.0 g'阿司匹林,置于2000 ml容量瓶中。接着,预先将1.0N盐酸加入到净化水中,并将PH调节至1.21。将该水溶液加入到容量瓶中的阿司匹林中,使总量为2,000ml。使用磁力搅拌器将Apyrin溶解在20℃的恒温水浴中。接下来,将由作为(II)中使用的原料的松木制成的药用煤研磨至-300目,加入其中的17g并搅拌3小时,分离上清液。吸附阿司匹林的药用木炭在25℃下用真空干燥器干燥12小时,置于密封的玻璃容器中,并在4℃的冰箱中保存。该样品用作胶囊的核心物质(下文缩写为ACASP)。后面将要描述使用高效液相色谱阿司匹林的药用碳的吸附量,从阿司匹林的上清液中的剩余量通过计算得到的结果,药用碳1克满足“每200毫克吸附量” / G'是的。
(2)微胶囊的制备
将乙基纤维素溶于250ml二氯甲烷中。乙基纤维素浓度设定为0.4,0.9,1.8,3.6%。接下来,在每种浓度下向每种乙基纤维素溶液中加入17g'ACASP,并用磁力搅拌器悬浮20分钟。为了制备胶囊,使用与前一部分相同的设备。首先,设置含有调整为25℃的恒温水浴4%明胶水溶液的容器中,该悬浮液中缓慢滴加,并将混合物逐渐加热至40℃与恒温水浴的温度。搅拌速度剥离乙基纤维素浓度在400rpm或更低0.4,0.9%,并调节,因为在所有的得到的胶囊将在反应容器的下部部分在500rpm或更小1.8,3.6%600转的情况下,沉降粗颗粒。24小时后,取出胶囊并用纯净水洗涤。在用真空干燥器在25℃下进一步干燥12小时后,将其紧密密封在玻璃容器中,置于冰箱中并在4℃下储存用于实验。
3。实验结果和讨论
(1)胶囊的制备条件
表VI-1显示了在每种制备条件下获得的胶囊的产率,内表面积,堆积密度等。随着乙基纤维素浓度的增加,内表面积趋于减少,并且产率显示出乙基纤维素浓度越高,产率越高的趋势。
|
乙基 纤维素(%) |
阿司匹林 吸附炭(%) |
搅拌速度 (rpm) |
收益率 (%) |
内表面积 (m 2 / g) |
体积密度 (g / ml) |
| 0.4 | 17 | 400 | 75.6 | 47.2 | 0.29 |
| 0.9 | 17 | 400 | 79.5 | 31.3 | 0.34 |
| 1.8 | 17 | 600 | 83.1 | 17.2 | 0.40 |
| 3.6 | 17 | 600 | 87.9 | 15.5 | 0.41 |
(2)溶出试验
图VII-1显示了在pH6.9的水溶液中作为实例,从胶囊洗脱的阿司匹林和水杨酸的溶出量与洗脱时间之间的关系。在pH 6.9时,阿司匹林和水杨酸几乎呈线性上升,直至洗脱时间为00.5至4小时,此后它几乎恒定。阿司匹林显示出比水杨酸更容易洗脱的倾向。而且,乙基纤维素浓度越低,洗脱越多,洗脱时间也趋于更快。在pH10.0时,阿司匹林的溶出量在显示最大值后降低,并且随着乙基纤维素浓度降低,显示最大值的洗脱时间短。相比于线性水杨酸的溶出量示出了后升高的恒定值,发现在许多情况下的乙基纤维素浓度的溶出量就越低。在pH 12时,只有水杨酸随着溶解时间的增加而线性上升,并且随着乙基纤维素浓度降低,洗脱时间更快并且洗脱量也更高。整体阿司匹林联系Yopi的总量从水杨酸在各pH的胶囊1克”溶解是在ACASP阿司匹林含量为约20〜23%,余量被认为保留在胶囊中。虽然还示出了ACASP的溶解测试结果,洗脱时间比达到近平衡的胶囊阿司匹林和水杨酸溶出量在约10分钟,是基本上相同的洗脱体积为胶囊的0.4%的乙基纤维素浓度我发现了。这似乎是胶囊中的药用炭的表面或吸附在孔中的阿司匹林的一部分被分解和洗脱。
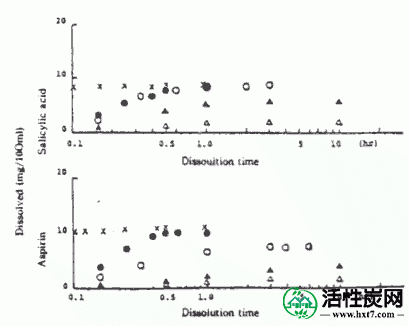
以上,药用炭吸附阿司匹林,乙基纤维素和壁膜,长洗脱时间阿司匹林联系Yopi水杨酸微囊,并发现乙基纤维素的浓度调节溶出量的可能性。也阿司匹林易发生时采取的,而肠吸收是期望的,并且当其被吸收信水杨酸钠,这个实验似乎是在这个意义上值得胃肠功能紊乱。我们预计,缓慢释放在制药领域添加严谨考虑,制备含有阿司匹林良好的除魔的微胶囊。
特点
这项研究有两个目标。首先,从活性炭工业的实际情况来看,我们正在扩大原料方面,特别是使用高度工业废料,我们已经从这些原料中建立了这样的制造方法。
另一个表明,由此获得的活性炭不仅仅用于工业用途或防止污染,而是用于更高质量的应用,例如用于药用煤,并且显示了其策略。
活性炭的制备,诀窍很多,但原料是更大的点不明确等,鉴于以上在本研究中,从层叠步骤非常基本的基本上是清楚的活性炭的制备。
本研究中,部分涉及到木材为主,因为从捡到为原料推测,其中由工业科学和技术机构(ITIT)的国际科研合作最初是带领菲律宾(NIST)国家研究所。其结果是,有可能得到有效的结果,并拿起了日本国际协力机构(JAICA)已建成在马尼拉的同一研究所的工业试验炉。在这种植物中,锯末是严重放电国家作为原料,碳化用作考虑到当地的条件平炉型。活化是流通式炉,其是内部加热型水蒸气活化。可以生产约20kg / hr的碳化物进料速率,3至7kg / hr的活性炭。获得到,该报告(II)中所描述的产品等同的,活化为1.0 N,用HCl,它肯定会适应作为药用碳。
生产活性炭。
使用活性炭进行化学制造。
活性炭的制造方法(特殊)1258469
活性炭的制造方法(特殊)1028524
吸附剂的制备方法(特殊)154628
缩写列表
| IP | lpil-ipil(イビルイビル) |
| 美联社 | Apitong(Apiton) |
| PA | 松(松) |
| IPC | Ipyrupil硬质合金 |
| APC | 硬质合金 |
| PAC | 松树碳化物 |
| IPCA | 来自ipyrupil碳化物的活化剂 |
| APCA | 从apiton的硬质合金中激活 |
| PACA | 松树碳水化合物的活化剂 |
| TA | Taiheiyo煤炭 |
| TAA | 太平洋煤比重分选木炭 |
| TAAS | 太平洋煤用硫酸处理 |
| TA 600 | Taiheiyo Coal 600°C硬质合金 |
| TAASA | TAAS的活动 |
| SN | Sunagawa煤炭 |
| ^ h | Horonai煤炭 |
| IB | Baiduri木炭 |
| H460 | Horonai木炭460°C碳化物 |
| S430 | Sunagawa煤炭430°C碳化物 |
| IBC | Baiduri木炭硬质合金 |
| IBSA | IBS的活动 |
| PVC | 聚氯乙烯 |
| PVDC | 聚偏二氯乙烯 |
| PE | 聚乙烯 |
| APP | Atasutake聚乙烯 |
| PANW | 纤维状聚丙烯腈共聚物 |
| PANPA | PANW在空气中热处理2小时 |
| PANBB | PANW在空气中热处理5小时 |
| PANC | 通过PANW在空气中在150,200和230℃下三个阶段热缩合获得的碳化物 |
| PVA | 聚乙烯醇 |
| PVAS | 硫酸处理的聚乙烯醇 |

Copyright © 2018 WWW.HXT7.COM 活性炭网 版权所有 桂ICP备15006904号-5
服务热线:13612777675 | 客服QQ:461763946 | 在线客服QQ: